![]()

![]()

Quickstart docs:
See Introduction to the Biolink datamodel slides for a background on the Biolink Model and its inception.
Conversion/validation code: https://github.com/NCATS-Tangerine/kgx
The purpose of the Biolink model is to provide a high level datamodel of biological entities (genes, diseases, phenotypes, pathways, individuals, substances, etc), their properties, relationships, and enumerate ways in which they can be associated.
The representation is independent of storage technology or metamodel (Solr documents, neo4j/property graphs, RDF/OWL, JSON, CSVs, etc). Different mappings to each of these are provided.
The specification of the reference Biolink model is a single YAML file following a custom meta-model. The basic elements of the YAML are:
This datamodel is being used in the NCATS Translator project. Not all these elements in the datamodel are used by the Translator; only a subset.
Protege view:

We divide these into relationship types (which connect two nodes together), node properties and edge properties.
See biolink json-ld context to find out more about identifier prefixes and their mappings.
Refer to mapping to neo4j for strategies on representing Biolink model in a Neo4j database.
Refer to mapping to RDF and RDF* for strategies on representing Biolink model in a RDF triple store.
The datamodel source is biolink-model.yaml. This is a YAML file that is intended to be relatively simple to view and edit in its native form.
The yaml definition is currently used to derive:
We leverage existing frameworks where possible. E.g json-schema allows codegen to other languages.
Additionally, this repo contains the metamodel definition of itself in YAML, together with code for working with datamodels. In theory this could be used in other domains but there is no plan for this at the moment.
See metamodel for details of the metamodel.
Prerequisites: Python 3.7+
To install,
make installIf you make changes to biolink-model.yaml then be sure to run the Makefile to generate up-to-date artifacts and documentation.
makeNote: the Makefile requires jsonschema2pojo.
If you are on a Mac, it can be installed using brew:
brew install jsonschema2pojoCurrently this is documented in the ingest artifacts repo, using non-computable cmap images:
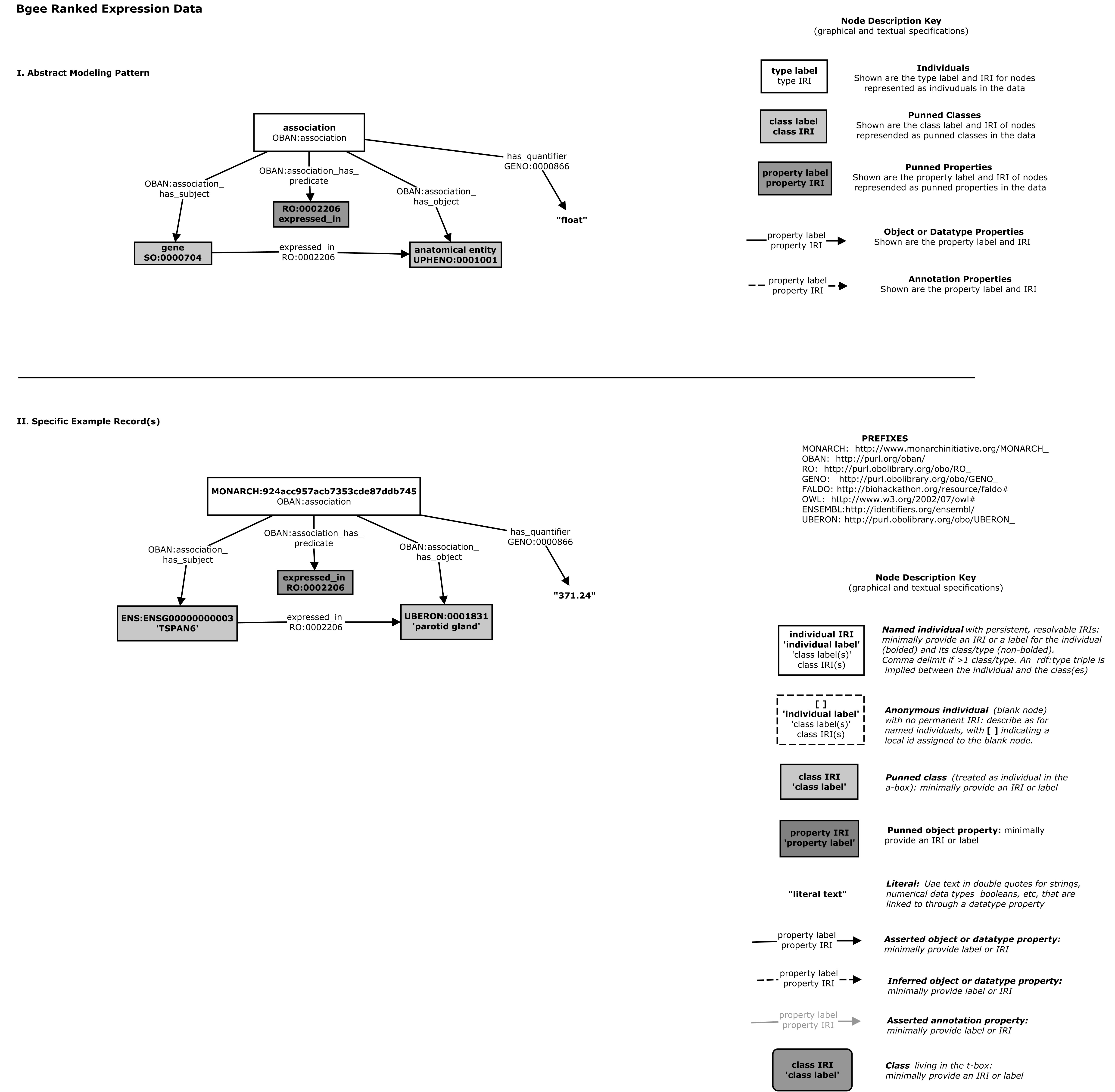
And also by the gene-anatomy cypher query which maps graphs conforming to the pattern to denormalized tuples for indexing in Solr
In the Biolink model this is explicitly represented using the gene to expression site association class definition
in the model
- name: gene to expression site association
is_a: association
description: >-
An association between a gene and an expression site, possibly qualified by stage/timing info
see_also: "https://github.com/monarch-initiative/ingest-artifacts/tree/master/sources/BGee"
slot_usage:
- slot: subject
type: gene or gene product
description: "gene in which variation is correlated with the phenotypic feature"
- slot: object
type: anatomical entity
description: "location in which the gene is expressed"
subclass_of: UBERON:0001062
examples:
- value: UBERON:0002037
description: cerebellum
- slot: relation
description: "expression relationship"
subproperty_of: "RO:0002206"
- slot: stage
type: developmental stage
description: "stage at which the gene is expressed in the site"
examples:
- value: UBERON:0000069
description: larval stage
- slot: quantifier
description: >-
can be used to indicate magnitude, or also rankingThis is used to generate various artifacts such as
Auto-generated UML diagram:

Auto-generated GraphQL definition:
type GeneToExpressionSiteAssociation {
qualifiers: [String]
stageQualifier: LifeStage
objectExtensions: [PropertyValuePair]
hasEvidence: String
publications: [Publication]
object: AnatomicalEntity!
hasEvidenceType: EvidenceType
hasEvidenceGraph: String
providedBy: Provider
label: String
relation: String!
negated: String
subject: GeneOrGeneProduct!
id: String!
quantifierQualifier: String
associationType: String
subjectExtensions: [PropertyValuePair]
}Auto-generated JSON Schema snippet:
"GeneToExpressionSiteAssociation": {
"description": "An association between a gene and an expression site, possibly qualified by stage/timing info. TBD: introduce subclasses for distinction between wild-type and experimental conditions?",
"properties": {
"association_type": {
"description": "connects an association to the type of association (e.g. gene to phenotype)",
"type": "string"
},
"has_evidence": {
"description": "connects an association to an instance of supporting evidence",
"type": "string"
},
"has_evidence_graph": {
"description": "connects an association to a graph object including a path from subject to object",
"type": "string"
},
"has_evidence_type": {
"description": "connects an association to the class of evidence used",
"type": "string"
},
"id": {
"type": "string"
},
"label": {
"description": "A human-readable name for a thing",
"type": "string"
},
"negated": {
"description": "if set to true, then the association is negated i.e. is not true",
"type": "string"
},
"object": {
"description": "connects an association to the object of the association. For example, in a gene-to-phenotype association, the gene is subject and phenotype is object.",
"type": "string"
},
"object_extensions": {
"description": "Additional relationships that are true of the object in the context of the association. For example, if the object is an anatomical term in an expression association, the object extensions may include part-of links",
"items": {
"type": "string"
},
"type": "array"
},
"provided_by": {
"description": "connects an association to the agent (person, organization or group) that provided it",
"type": "string"
},
"publications": {
"description": "connects an association to publications supporting the association",
"items": {
"type": "string"
},
"type": "array"
},
"qualifiers": {
"description": "connects an association to qualifiers that modify or qualify the meaning of that association",
"items": {
"type": "string"
},
"type": "array"
},
"quantifier_qualifier": {
"description": "A measurable quantity for the object of the association",
"type": "string"
},
"relation": {
"description": "the relationship type by which a subject is connected to an object in an association",
"type": "string"
},
"stage_qualifier": {
"description": "stage at which expression takes place",
"type": "string"
},
"subject": {
"description": "connects an association to the subject of the association. For example, in a gene-to-phenotype association, the gene is subject and phenotype is object.",
"type": "string"
},
"subject_extensions": {
"description": "Additional relationships that are true of the subject in the context of the association. For example, if the subject is a gene product in a functional association, the subject extensions may represent an isoform or a specific post-translational state",
"items": {
"type": "string"
},
"type": "array"
}
},
"required": [],
"title": "GeneToExpressionSiteAssociation",
"type": "object"
},Why invent our own yaml and not use JSON-Schema, SQL, UML, ProtoBuf, OWL, etc.
Each of these is tied to a particular formalisms, e.g. JSON Schema to trees, OWL to open world logic. There are various impedance mismatches in converting between these. The goal was to develop something simple and more general that is not tied to any one serialization format or set of assumptions.
There are other projects with similar goals, e.g Schema Salad
It may be possible to align with these.
Here X may be BioSchemas, some upper ontology (BioTop), UMLS metathesaurus, bio*, various other attempts to model all of biology in an object model.
Currently as far as we know there is no existing reference datamodel that is flexible enough to be used here.
For operations such as CURIE lookup, finding class by synonyms, get parents, get ancestors, etc. please make use of biolink-model-toolkit. It provides a convenience methods for traversing Biolink Model.